Deep Learning
Docking Proteins to Deny Disease: Computational Considerations for Simulating Protein-Ligand Interaction
December 17, 2020
19 min read

Molecular docking is a powerful tool for studying biological macromolecules and their binding partners. This amounts to a complex and varied field of study, but of particular interest to the drug discovery industry is the computational study of interactions between physiologically important proteins and small molecules.
This is an effective way to search for small molecule drug candidates that bind to a native human protein, either attenuating or enhancing activity to ameliorate some disease condition. Or, it could mean binding to and blocking a protein used by pathogens like viruses or bacteria to hijack human cells.
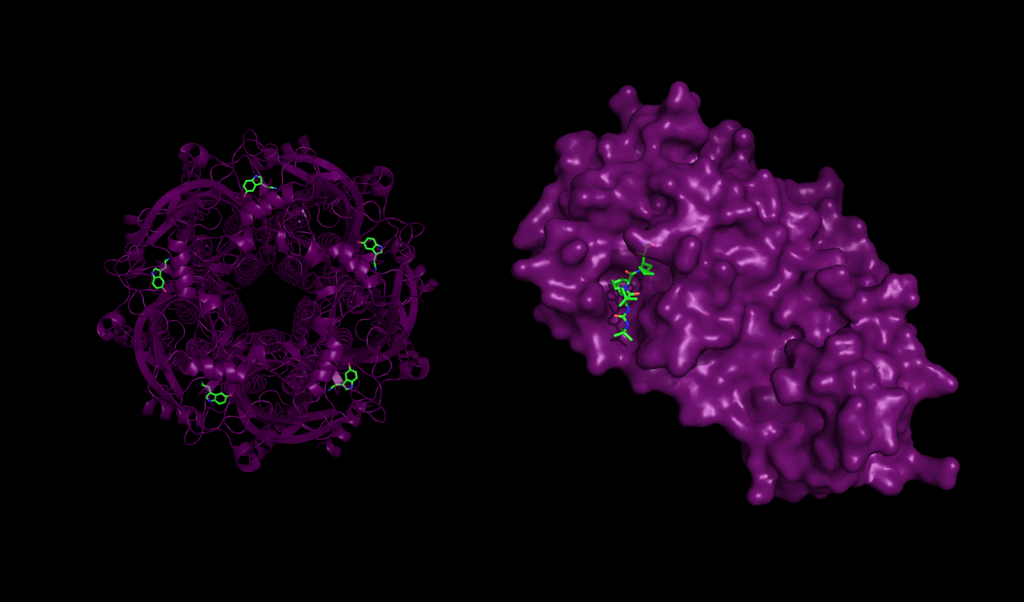
Protein-ligand interaction in health and disease. Left: 5-HT3 receptor with serotonin molecules bound, an interaction that plays important roles in the central nervous systems. Right: Mpro, a major protease of the SARS-CoV-2 coronavirus responsible for COVID-19, bound to the inhibitor Boceprevir.
Drug discovery has come a long way from its inception. The first drugs used to treat maladies were probably discovered in prehistory in the same way as the first recreational drugs were: someone noticing strange effects after ingesting plant or animal matter out of boredom, hunger, or curiosity. This knowledge would have spread rapidly by word of mouth and observation. Even orangutans have been observed producing and using medicine by chewing leaves into a foamy lather to use as a topical painkiller. The practice is local to orangutans in only a few places in Borneo, so it was almost certainly discovered and passed on rather than as a result of instinctual use, which is actually quite common even among invertebrates.
Historical drug discovery from the times of ancient Sumerians to the Renaissance in Europe was also a matter of observation and guesswork. This persisted as the main method for drug discovery well into the 20th century. Many of the most impactful modern medicines were discovered by observation and happenstance, including vaccines, discovered by Edward Jenner in the 1700s after observing dairy maids that were immune to smallpox thanks to exposure to the bovine version of the disease, and antibiotics, discovered by Alexander Fleming as a side-effect of keeping a somewhat messy lab.
Discovery by observation provides hints at where to look, and researchers and a burgeoning pharmacology industry began to systematically isolate compounds from nature to screen them for drug activity, especially after World War II. But it’s easier to isolate (and later synthesize) large libraries of chemical compounds than it is to test each one in a cell culture or animal model for every possible disease. This has culminated in many pharmaceutical companies maintaining vast libraries of small molecule compounds with likely physiological activity. As a result, choosing which compounds out of thousands are worth studying more closely to combat a given disease, known as lead generation, is the first hurdle to leap in the modern drug discovery process. Automated high-throughput screening is a promising tool for discovering leads, at least when it’s done intelligently. Computational chemistry, on the other hand, has the potential to be vastly more efficient than any sort of screening you can do in a wet lab.
There are two main categories docking simulations can fall under: static/rigid, versus flexible/soft.
Conceptually these are related to two mental models of how proteins interact with other molecules, including other proteins: the lock and key model and the induced fit model. The latter is sometimes referred to as the “glove” model, relating to the way that a latex glove stretches to accommodate and take the shape of a hand. These aren’t so much competing models as they are different levels of abstraction. Proteins are almost never going to be perfectly rigid under physiological temperatures and concentrations.
From our computational perspective, flexible docking is closer to actual conditions experienced by proteins and ligands, but rigid docking will clearly be much faster to compute. A useful compromise between speed and precision is to screen against several different static structural conformations of the same protein. That being said, it’s hard to get all of the details right in a more complex simulation such as flexible docking, and in those cases it’s better to be less precise but more accurate.
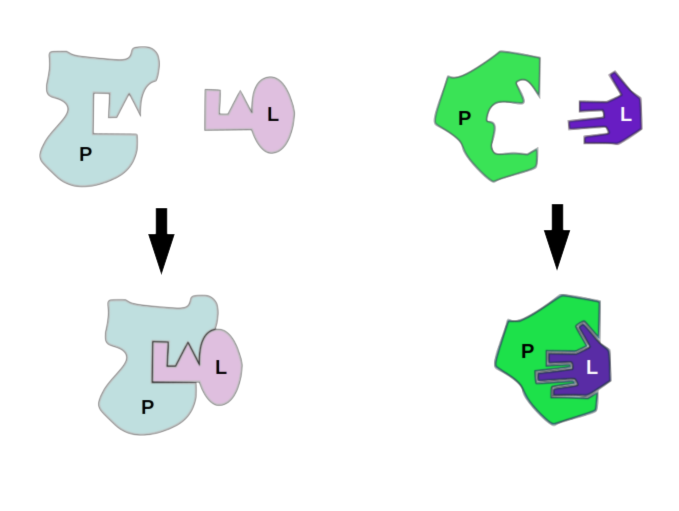
Models of protein-ligand interaction, lock-and-key interaction and induced fit, or “glove” binding. These ideas about protein binding are parallelled in rigid and flexible docking simulation, respectively.
Protein-ligand docking as a means for lead generation has many parallels to deep learning. The technique has taken on a qualitatively different character over the last few decades, thanks to Moore’s law and related drivers of readily available computational power.
Although it’s widely accepted that Moore’s law isn’t really active anymore, at least in terms of the number of transistors that can be packed onto a given piece of silicon, the development of parallel computing in hardware accelerators like general-purpose graphics processing units (GP-GPUs), field programmable gate arrays (FPGAs), and application specific integrated circuits (ASICs) has ensured that we still see impressive yearly improvements in computation per Watt or computation per dollar.
As we’ll discuss in this post, some molecular docking software programs have started to take advantage of GPU computation, and hybrid methodologies that implement molecular docking and deep learning inference side by side can speed up virtual screening by more than 2 orders of magnitude.
Even without explicit support for parallelization, modern computation has driven the adoption of protein-ligand docking for high throughput virtual screening. Docking is one of several strategies for screening that include predicting quantitative structure-activity relationship (QSAR) and virtual screening based on chemical characteristics and metrics associated with a large library.
Simulated protein-ligand docking has historically been used in a more focused manner. Until recently, you’d be more likely to perform a docking study with only one or a handful of ligands. This generally entails a far amount of manual ligand processing as well as direct inspection of the protein-ligand interaction, and would be conducted at the hands of a human operator with a huge degree of technical knowledge (or as it says in the non-compete in your contract proprietary “know-how”).
Interested in getting faster results?
Learn more about Exxact AI workstations starting at $3,700
These days it’s much more feasible to screen hundreds to thousands of small molecules against a handful of different conformations of the same protein, and it’s considered good practice to use a few different docking programs as well, as they all have their own idiosyncrasies. With each docking simulation typically taking on the order of seconds (for one ligand on one CPU), this can take a significant amount of time and compute even when spread out across 64 or so CPU cores. For large screens, it pays to use a program that can distribute the workload over several machines or nodes in a cluster.
| Software | Open Source? | License | 1st Year Published | GPU Support? | Multicore
CPU Support? |
| Autodock | yes | GNU GPL | 1990 | yes | no |
| Vina | yes | Apache | 2010 | no | yes |
| Smina | yes | Apache | 2012 | no | yes |
| rDock | Yes
(as of 2012) | GNU LGPL | 1998
Open-sourced in 2012 | no | yes |
| RxDock | yes | GNU LGPL | 2019 | no | yes
support for an “unlimited number of CPUs” |
| CABS-Dock | yes | MIT | 2015 | no | no |
| RosettaLigand | no | Academic/
Commercial | 2006 | no | for ligand preparation |
| OEDocking & FRED | no | Academic/
Government/ Commercial | 2011 | no | yes |
| Molegro Virtual Docker | no | Commercial | 2006 | yes | not clear |
| ICM-Pro | no | Commercial | 1994 | no | no |
| Hex | no | Academic/
Government/ Commercial | ~1998 | yes | yes |
| Schrödinger Glide | no | Academic/
Commercial | 2004 | no | likely |
Non-exhaustive list of software for protein-ligand docking simulation.
We can see in the table above that very few docking programs have been designed or modified to take advantage of the rapid progress in GPU capabilities. In that regard molecular docking trails deep learning by 6 years or so.
Autodock-GPU (open source option), Molegro Virtual Docker, and Hex are the only examples in the table that have GPU support. Of those, Autodock-GPU is the only open source option, Hex has a free academic license, and Molegro Virtual Docker doesn’t list pricing for the license on the website. Autodock 4 has also been implemented to take advantage of FPGAs as well, so if your HPC capabilities include FPGAs they don’t have to go to waste.
Even if using a docking program that doesn’t support GPU computation, there are other ways to take advantage of hardware acceleration by combining physics-based docking simulation with deep learning, and other machine learning models.
Even if most molecular docking software doesn’t readily support GPU acceleration, there’s an easy and straightforward way to take advantage of GPUs and all of the technical development in GPU computing that has been spurred on by the rising value proposition of deep learning, and that’s to find a place in your workflow where you actually can plug in some deep learning.
Neural networks of even modest depth are technically capable of universal function approximation, and researchers have shown their utility in approximating many physical phenomena at various scales. Deep NNs can provide shortcuts by learning relationships between structures and molecule activity directly, but they can also work together with a molecular docking program to make high throughput screening more efficient.
Austin Clyde describes a deep learning pre-filtering technique for high throughput virtual screening using a modified 50 layer ResNet neural network. In this scheme, the neural network predicts which ligands (out of a chemical library of more than 100 million compounds) are more likely to demonstrate strong interaction with the target protein. Those chosen ligands then enter the molecular docking pipeline to be docked with AutoDock-GPU. This vastly decreases the ligand search space while still discovering the majority of the best lead candidates.
Remarkably the ResNet model is trained to predict the best docking candidates based on a 2D image of each small molecule, essentially replacing the human intuition that might go into choosing candidates for screening.
The optimal use of available compute depends on the relationship between the GPU(s) and the number of CPU cores on the machine used for screening. To fully take advantage of the GPU (running ResNet) and the CPU cores (orchestrating docking) the researchers adjust the decision threshold to screen out different proportions of the molecular library. Clyde gave examples: screening only the top 1% of molecules chosen by the ResNet retains 50% of the true top 1% and 70% of the top 0.05% of molecules, and screening the top 10% chosen by ResNet recovers all of the top 0.1%. In some cases Clyde describes a speedup of 130x over naively screening all candidates. A technical description of this project, denoted IMPECCABLE (an acronym that is surely trying too hard), is available on Arxiv.
We looked at 2 open source molecular docking programs, AutoDock-GPU and Smina.
While these are both in the AutoDock lineage (Autodock Vina succeeds AutoDock, and Smina is a fork of Vina with emphasis on custom scoring functions), they have different computational strengths. AutoDock-GPU, as you can guess, supports running its “embarrassingly parallel” Lamarckian Genetic Algorithm on the GPU, while Smina can take good advantage of multiple cpu cores up to the value assigned by the –exhaustiveness flag.
Smina uses a different algorithm, the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm so the comparison isn’t direct, but the run speeds are quite comparable using the default settings. Increasing the thoroughness of search by setting -nrun (used by AutoDock-GPU) and –exhaustiveness (used by Smina) flags to high values, however, favors the multicore Smina over the GPU-enabled AutoDock.
Smina is available for download as a static binary on Sourceforge, or you can download and build the source code from Github. You’ll have to make AutoDock-GPU from source if you want to follow along, following the instructions from their Github repository. You’ll need either CUDA or OpenCL installed, and make sure to pay attention to environmental variables you’ll need to parametrize the build: DEVICE, NUMWI, GPU_LIBRARY_PATH and GPU_INCLUDE_PATH.
We used PyMol, openbabel and this script to prepare the pdb structure 1gpk for molecular docking with Smina. Preparation for AutoDock-GPU was a little more involved, as it relies on building a grid with autodock4, part of AutoDockTools. Taking inspiration from this thread on bioinformatics stack exchange, and taking the .pdbqt files prepared for docking with Smina as a starting point, you can use the following commands to prepare the map and grid files for AutoDock:
| cd <path_to_MGLTools>/MGLToolsPckgs/AutoDockTools/Utilities24/
# after placing your pdbqt files in the Utilities24 folder # NB You’ll need to use the included pythonsh version of python, # and you may have to reinstall NumPy for it! ../../../bin/pythonsh prepare_gpf4.py -l 1gpk-hup.pdbqt -r 1gpk-receptor.pdbqt -y autogrid4 -p 4oo8-cas9.gpf |
That should give the files you need to run AutoDock-GPU, but it can be a pain to install all the AutoDockTools on top of building AutoDock-GPU in the first place, so feel free to download the example files here.
To run docking with Smina, cd to the directory with the Smina binary and your pdbqt files for docking, then run:
| ./smina.static -r ./1gpk-receptor.pdbqt -l ./1gpk-hup.pdbqt –autobox_ligand ./1gpk-hup.pdbqt \
–autobox_add 8 -o ./output_file.pdbqt |
And you should get some results that look similar to:
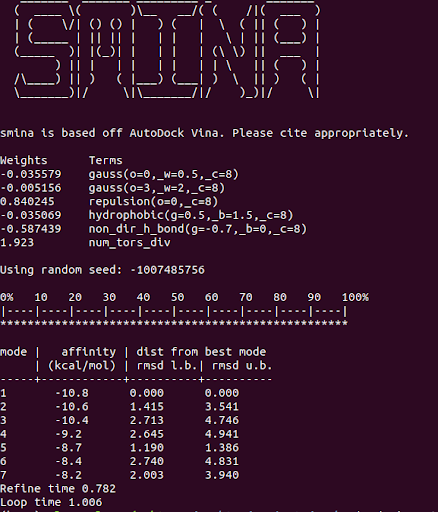
One advantage of Smina over Vina is the convenience flags for autoboxing your ligand. This allows you to automatically define the docking search space based on a ligand structure that was removed from a known binding pocket on your protein. Vina requires you to instead define the x,y,z, center coordinates and search volume.
Smina achieved a docking runtime of just over 1 second on 1 CPU for the 1gpk structure, with the default exhaustiveness of 1. Let’s see if AutoDock-GPU can do any better:
| ./bin/autodock_gpu_128wi -ffile ./1gpk-receptor.maps.fld \
-lfile 1gpk-hup.pdbqt |
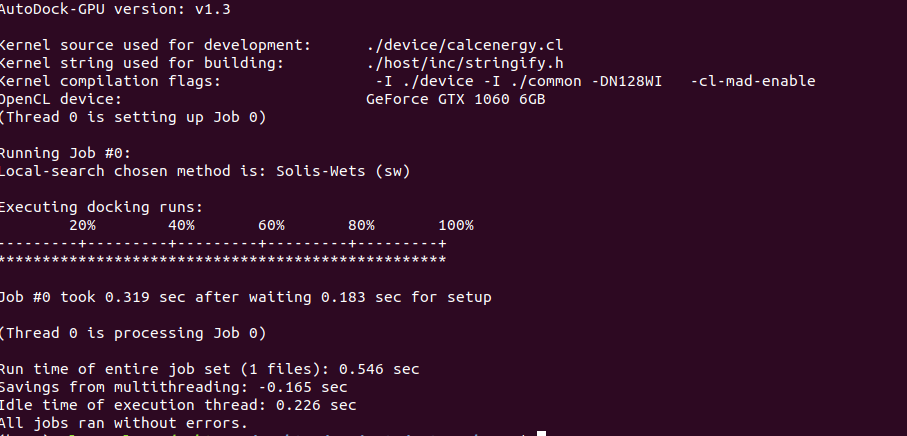
Note that your autodock executable may have a slightly different filename if you used different parameters when building. We can see that AutoDock-GPU was slightly faster than Smina in this example, almost by a factor of 2 in fact. But if we want to run docking with more precision by expanding the search, we can ramp up the -nrun and –exhaustiveness flags for some surprising results.
| ./smina.static -r ./1gpk-receptor.pdbqt -l ./1gpk-hup.pdbqt –autobox_ligand ./1gpk-hup.pdbqt \
–autobox_add 8 –exhaustiveness 1000 \ -o ./output_file.pdbqt |
Running smina with exhaustiveness of 1000 gives us a very satisfying CPU usage that looks great when visualized with htop, and a runtime of about 20 seconds.
If we do something similar with AutoDock-GPU and the -nrun flag:
| ./bin/autodock_gpu_128wi -ffile ./1gpk-receptor.maps.fld \
-lfile 1gpk-hup.pdbqt -nrun 1000 |
We get a runtime of just over 2 minutes. As mentioned earlier, accuracy is more important than precision when it comes to finding good drug candidates, and using 1000 for -nrun or –exhaustiveness is a truly exaggerated and preposterous way to set up your docking run (values around 8 are much more reasonable). Nonetheless, it may be more beneficial to adopt a complementary hybrid approach for high-throughput virtual screening, running docking on multiple CPU cores and using deep learning and GPUs to filter candidates, as described above for the IMPECCABLE pipeline.
In any case, speed doesn’t matter if we can’t trust the results so it pays to visually inspect docking results every once in a while.
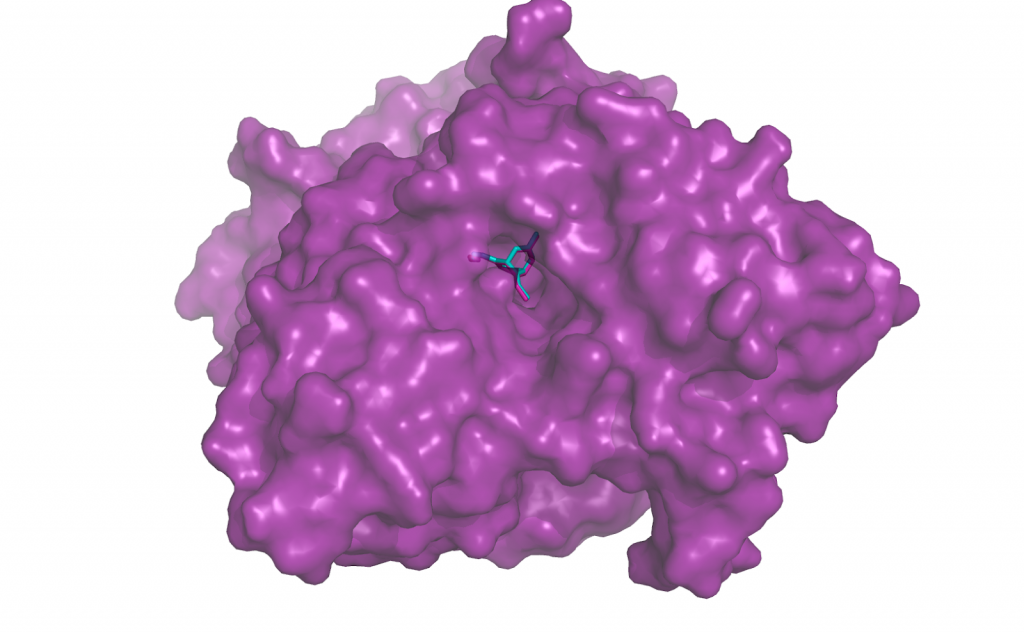
Re-docking Huperzine to acetylcholinesterase (PDB ID 1gpk). The turquoise small molecule is the output from docking program Smina. The re-docked position overlaps substantially with the true ligand position from the solved structure, in pink.
Hopefully this whirlwind tour of an article has helped to pique your interest in molecular docking. And now you can see how high throughput virtual screening can take advantage of modern multicore CPUs and GPU accelerators to blaze through vast compound libraries.
This application of compute to find chemical candidates for drugs in a targeted manner, especially when it’s done with a clever combination of machine learning and algorithmic simulation, has the potential to find new remedies faster, with fewer side-effects and better efficacy.
Molecular docking is a powerful tool for studying biological macromolecules and their binding partners. This amounts to a complex and varied field of study, but of particular interest to the drug discovery industry is the computational study of interactions between physiologically important proteins and small molecules.
This is an effective way to search for small molecule drug candidates that bind to a native human protein, either attenuating or enhancing activity to ameliorate some disease condition. Or, it could mean binding to and blocking a protein used by pathogens like viruses or bacteria to hijack human cells.
Protein-ligand interaction in health and disease. Left: 5-HT3 receptor with serotonin molecules bound, an interaction that plays important roles in the central nervous systems. Right: Mpro, a major protease of the SARS-CoV-2 coronavirus responsible for COVID-19, bound to the inhibitor Boceprevir.
Drug discovery has come a long way from its inception. The first drugs used to treat maladies were probably discovered in prehistory in the same way as the first recreational drugs were: someone noticing strange effects after ingesting plant or animal matter out of boredom, hunger, or curiosity. This knowledge would have spread rapidly by word of mouth and observation. Even orangutans have been observed producing and using medicine by chewing leaves into a foamy lather to use as a topical painkiller. The practice is local to orangutans in only a few places in Borneo, so it was almost certainly discovered and passed on rather than as a result of instinctual use, which is actually quite common even among invertebrates.
Historical drug discovery from the times of ancient Sumerians to the Renaissance in Europe was also a matter of observation and guesswork. This persisted as the main method for drug discovery well into the 20th century. Many of the most impactful modern medicines were discovered by observation and happenstance, including vaccines, discovered by Edward Jenner in the 1700s after observing dairy maids that were immune to smallpox thanks to exposure to the bovine version of the disease, and antibiotics, discovered by Alexander Fleming as a side-effect of keeping a somewhat messy lab.
Discovery by observation provides hints at where to look, and researchers and a burgeoning pharmacology industry began to systematically isolate compounds from nature to screen them for drug activity, especially after World War II. But it’s easier to isolate (and later synthesize) large libraries of chemical compounds than it is to test each one in a cell culture or animal model for every possible disease. This has culminated in many pharmaceutical companies maintaining vast libraries of small molecule compounds with likely physiological activity. As a result, choosing which compounds out of thousands are worth studying more closely to combat a given disease, known as lead generation, is the first hurdle to leap in the modern drug discovery process. Automated high-throughput screening is a promising tool for discovering leads, at least when it’s done intelligently. Computational chemistry, on the other hand, has the potential to be vastly more efficient than any sort of screening you can do in a wet lab.
There are two main categories docking simulations can fall under: static/rigid, versus flexible/soft.
Conceptually these are related to two mental models of how proteins interact with other molecules, including other proteins: the lock and key model and the induced fit model. The latter is sometimes referred to as the “glove” model, relating to the way that a latex glove stretches to accommodate and take the shape of a hand. These aren’t so much competing models as they are different levels of abstraction. Proteins are almost never going to be perfectly rigid under physiological temperatures and concentrations.
From our computational perspective, flexible docking is closer to actual conditions experienced by proteins and ligands, but rigid docking will clearly be much faster to compute. A useful compromise between speed and precision is to screen against several different static structural conformations of the same protein. That being said, it’s hard to get all of the details right in a more complex simulation such as flexible docking, and in those cases it’s better to be less precise but more accurate.
Models of protein-ligand interaction, lock-and-key interaction and induced fit, or “glove” binding. These ideas about protein binding are parallelled in rigid and flexible docking simulation, respectively.
Protein-ligand docking as a means for lead generation has many parallels to deep learning. The technique has taken on a qualitatively different character over the last few decades, thanks to Moore’s law and related drivers of readily available computational power.
Although it’s widely accepted that Moore’s law isn’t really active anymore, at least in terms of the number of transistors that can be packed onto a given piece of silicon, the development of parallel computing in hardware accelerators like general-purpose graphics processing units (GP-GPUs), field programmable gate arrays (FPGAs), and application specific integrated circuits (ASICs) has ensured that we still see impressive yearly improvements in computation per Watt or computation per dollar.
As we’ll discuss in this post, some molecular docking software programs have started to take advantage of GPU computation, and hybrid methodologies that implement molecular docking and deep learning inference side by side can speed up virtual screening by more than 2 orders of magnitude.
Even without explicit support for parallelization, modern computation has driven the adoption of protein-ligand docking for high throughput virtual screening. Docking is one of several strategies for screening that include predicting quantitative structure-activity relationship (QSAR) and virtual screening based on chemical characteristics and metrics associated with a large library.
Simulated protein-ligand docking has historically been used in a more focused manner. Until recently, you’d be more likely to perform a docking study with only one or a handful of ligands. This generally entails a far amount of manual ligand processing as well as direct inspection of the protein-ligand interaction, and would be conducted at the hands of a human operator with a huge degree of technical knowledge (or as it says in the non-compete in your contract proprietary “know-how”).
Interested in getting faster results?
Learn more about Exxact AI workstations starting at $3,700
These days it’s much more feasible to screen hundreds to thousands of small molecules against a handful of different conformations of the same protein, and it’s considered good practice to use a few different docking programs as well, as they all have their own idiosyncrasies. With each docking simulation typically taking on the order of seconds (for one ligand on one CPU), this can take a significant amount of time and compute even when spread out across 64 or so CPU cores. For large screens, it pays to use a program that can distribute the workload over several machines or nodes in a cluster.
| Software | Open Source? | License | 1st Year Published | GPU Support? | Multicore
CPU Support? |
| Autodock | yes | GNU GPL | 1990 | yes | no |
| Vina | yes | Apache | 2010 | no | yes |
| Smina | yes | Apache | 2012 | no | yes |
| rDock | Yes
(as of 2012) | GNU LGPL | 1998
Open-sourced in 2012 | no | yes |
| RxDock | yes | GNU LGPL | 2019 | no | yes
support for an “unlimited number of CPUs” |
| CABS-Dock | yes | MIT | 2015 | no | no |
| RosettaLigand | no | Academic/
Commercial | 2006 | no | for ligand preparation |
| OEDocking & FRED | no | Academic/
Government/ Commercial | 2011 | no | yes |
| Molegro Virtual Docker | no | Commercial | 2006 | yes | not clear |
| ICM-Pro | no | Commercial | 1994 | no | no |
| Hex | no | Academic/
Government/ Commercial | ~1998 | yes | yes |
| Schrödinger Glide | no | Academic/
Commercial | 2004 | no | likely |
Non-exhaustive list of software for protein-ligand docking simulation.
We can see in the table above that very few docking programs have been designed or modified to take advantage of the rapid progress in GPU capabilities. In that regard molecular docking trails deep learning by 6 years or so.
Autodock-GPU (open source option), Molegro Virtual Docker, and Hex are the only examples in the table that have GPU support. Of those, Autodock-GPU is the only open source option, Hex has a free academic license, and Molegro Virtual Docker doesn’t list pricing for the license on the website. Autodock 4 has also been implemented to take advantage of FPGAs as well, so if your HPC capabilities include FPGAs they don’t have to go to waste.
Even if using a docking program that doesn’t support GPU computation, there are other ways to take advantage of hardware acceleration by combining physics-based docking simulation with deep learning, and other machine learning models.
Even if most molecular docking software doesn’t readily support GPU acceleration, there’s an easy and straightforward way to take advantage of GPUs and all of the technical development in GPU computing that has been spurred on by the rising value proposition of deep learning, and that’s to find a place in your workflow where you actually can plug in some deep learning.
Neural networks of even modest depth are technically capable of universal function approximation, and researchers have shown their utility in approximating many physical phenomena at various scales. Deep NNs can provide shortcuts by learning relationships between structures and molecule activity directly, but they can also work together with a molecular docking program to make high throughput screening more efficient.
Austin Clyde describes a deep learning pre-filtering technique for high throughput virtual screening using a modified 50 layer ResNet neural network. In this scheme, the neural network predicts which ligands (out of a chemical library of more than 100 million compounds) are more likely to demonstrate strong interaction with the target protein. Those chosen ligands then enter the molecular docking pipeline to be docked with AutoDock-GPU. This vastly decreases the ligand search space while still discovering the majority of the best lead candidates.
Remarkably the ResNet model is trained to predict the best docking candidates based on a 2D image of each small molecule, essentially replacing the human intuition that might go into choosing candidates for screening.
The optimal use of available compute depends on the relationship between the GPU(s) and the number of CPU cores on the machine used for screening. To fully take advantage of the GPU (running ResNet) and the CPU cores (orchestrating docking) the researchers adjust the decision threshold to screen out different proportions of the molecular library. Clyde gave examples: screening only the top 1% of molecules chosen by the ResNet retains 50% of the true top 1% and 70% of the top 0.05% of molecules, and screening the top 10% chosen by ResNet recovers all of the top 0.1%. In some cases Clyde describes a speedup of 130x over naively screening all candidates. A technical description of this project, denoted IMPECCABLE (an acronym that is surely trying too hard), is available on Arxiv.
We looked at 2 open source molecular docking programs, AutoDock-GPU and Smina.
While these are both in the AutoDock lineage (Autodock Vina succeeds AutoDock, and Smina is a fork of Vina with emphasis on custom scoring functions), they have different computational strengths. AutoDock-GPU, as you can guess, supports running its “embarrassingly parallel” Lamarckian Genetic Algorithm on the GPU, while Smina can take good advantage of multiple cpu cores up to the value assigned by the –exhaustiveness flag.
Smina uses a different algorithm, the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm so the comparison isn’t direct, but the run speeds are quite comparable using the default settings. Increasing the thoroughness of search by setting -nrun (used by AutoDock-GPU) and –exhaustiveness (used by Smina) flags to high values, however, favors the multicore Smina over the GPU-enabled AutoDock.
Smina is available for download as a static binary on Sourceforge, or you can download and build the source code from Github. You’ll have to make AutoDock-GPU from source if you want to follow along, following the instructions from their Github repository. You’ll need either CUDA or OpenCL installed, and make sure to pay attention to environmental variables you’ll need to parametrize the build: DEVICE, NUMWI, GPU_LIBRARY_PATH and GPU_INCLUDE_PATH.
We used PyMol, openbabel and this script to prepare the pdb structure 1gpk for molecular docking with Smina. Preparation for AutoDock-GPU was a little more involved, as it relies on building a grid with autodock4, part of AutoDockTools. Taking inspiration from this thread on bioinformatics stack exchange, and taking the .pdbqt files prepared for docking with Smina as a starting point, you can use the following commands to prepare the map and grid files for AutoDock:
| cd <path_to_MGLTools>/MGLToolsPckgs/AutoDockTools/Utilities24/
# after placing your pdbqt files in the Utilities24 folder # NB You’ll need to use the included pythonsh version of python, # and you may have to reinstall NumPy for it! ../../../bin/pythonsh prepare_gpf4.py -l 1gpk-hup.pdbqt -r 1gpk-receptor.pdbqt -y autogrid4 -p 4oo8-cas9.gpf |
That should give the files you need to run AutoDock-GPU, but it can be a pain to install all the AutoDockTools on top of building AutoDock-GPU in the first place, so feel free to download the example files here.
To run docking with Smina, cd to the directory with the Smina binary and your pdbqt files for docking, then run:
| ./smina.static -r ./1gpk-receptor.pdbqt -l ./1gpk-hup.pdbqt –autobox_ligand ./1gpk-hup.pdbqt \
–autobox_add 8 -o ./output_file.pdbqt |
And you should get some results that look similar to:
One advantage of Smina over Vina is the convenience flags for autoboxing your ligand. This allows you to automatically define the docking search space based on a ligand structure that was removed from a known binding pocket on your protein. Vina requires you to instead define the x,y,z, center coordinates and search volume.
Smina achieved a docking runtime of just over 1 second on 1 CPU for the 1gpk structure, with the default exhaustiveness of 1. Let’s see if AutoDock-GPU can do any better:
| ./bin/autodock_gpu_128wi -ffile ./1gpk-receptor.maps.fld \
-lfile 1gpk-hup.pdbqt |
Note that your autodock executable may have a slightly different filename if you used different parameters when building. We can see that AutoDock-GPU was slightly faster than Smina in this example, almost by a factor of 2 in fact. But if we want to run docking with more precision by expanding the search, we can ramp up the -nrun and –exhaustiveness flags for some surprising results.
| ./smina.static -r ./1gpk-receptor.pdbqt -l ./1gpk-hup.pdbqt –autobox_ligand ./1gpk-hup.pdbqt \
–autobox_add 8 –exhaustiveness 1000 \ -o ./output_file.pdbqt |
Running smina with exhaustiveness of 1000 gives us a very satisfying CPU usage that looks great when visualized with htop, and a runtime of about 20 seconds.
If we do something similar with AutoDock-GPU and the -nrun flag:
| ./bin/autodock_gpu_128wi -ffile ./1gpk-receptor.maps.fld \
-lfile 1gpk-hup.pdbqt -nrun 1000 |
We get a runtime of just over 2 minutes. As mentioned earlier, accuracy is more important than precision when it comes to finding good drug candidates, and using 1000 for -nrun or –exhaustiveness is a truly exaggerated and preposterous way to set up your docking run (values around 8 are much more reasonable). Nonetheless, it may be more beneficial to adopt a complementary hybrid approach for high-throughput virtual screening, running docking on multiple CPU cores and using deep learning and GPUs to filter candidates, as described above for the IMPECCABLE pipeline.
In any case, speed doesn’t matter if we can’t trust the results so it pays to visually inspect docking results every once in a while.
Re-docking Huperzine to acetylcholinesterase (PDB ID 1gpk). The turquoise small molecule is the output from docking program Smina. The re-docked position overlaps substantially with the true ligand position from the solved structure, in pink.
Hopefully this whirlwind tour of an article has helped to pique your interest in molecular docking. And now you can see how high throughput virtual screening can take advantage of modern multicore CPUs and GPU accelerators to blaze through vast compound libraries.
This application of compute to find chemical candidates for drugs in a targeted manner, especially when it’s done with a clever combination of machine learning and algorithmic simulation, has the potential to find new remedies faster, with fewer side-effects and better efficacy.



